
")
By Jayet Moon and Arun Mathew

In early July, FDA released a new draft guidance entitled “Purpose and Content of Use-Based Risk Analyses for Drugs, Biological Products, and Combination Products.” It provides guidance to industry and FDA staff on the purpose and content of a usability risk management tool, a use-based risk analysis (URRA), which has recently gained significant traction at FDA. A number of FDA guidance documents since 2016 that reference the URRA express FDA’s expectation (read: recommendation) to not only have a URRA as part of the premarket submission, but also to update it as needed after market launch. In the premarket phase, a well-crafted URRA is intended to help identify human factors data needs during product development and support a regulatory filing.
While these guidelines explicitly apply to drug-biologic combination products that contain a device component, the concepts contained therein are readily applicable to medical devices, in vitro devices, and in some cases to stand-alone drugs and biological products, such as when prescription and non-prescription drugs/biological products for humans are part of an NDA, BLA, or IND (including their supplements).
This is the FDA’s concise, step-by-step guidance on URRA so far, and it reads almost like a work instruction.
FDA recommends that sponsors contact FDA early in the design and development phase of a product to discuss the human factors (HR) development program through meeting requests.
That what?
The URRA is a risk management tool used to identify usage-related hazards associated with product use and the actions taken to reduce those risks. The URRA supports the entire RF engineering process and should be considered as part of a comprehensive risk management framework in accordance with ISO 14971 or ICH Q9, whichever is appropriate.
The why?
URRA is the tool for identifying use-related hazards associated with the combination product user interface design and characterizing risks so that they can be mitigated (e.g., through risk controls) or eliminated through improved product user interface design. It helps determine whether the results of an RF validation study are justified as part of a new premarket application. In addition, URRA is a key component in developing an RF validation study protocol and informing the acceptance of residual risks. URRA can also help demonstrate compliance with applicable requirements codified in 21 CFR Part 4 Subpart A for combination products.
The when?
Unlike a failure mode and effects analysis (uFMEA), which has traditionally been started later in the system-level design process (often after design inputs and outputs are close to being locked in), the URRA is started early during the product development process and healthy use of the URRA is expected to generate new user, design, and user interface (UI) requirements, some of which could be part of the application of risk control measures. To be clear, in this guidance, FDA expects that the uFMEA can be one of the tools to identify potential application failures.
FDA expects the URRA to be used throughout all phases of the product life cycle and to be updated as product design changes or new risks are identified during development or after market launch.
According to the FDA, the URRA should contain:
- A comprehensive list of all tasks required to use the product
- The possible usage errors and damage that may occur during these tasks
- A determination of whether each task is a critical task
- Risk controls in user interface design to reduce usage errors
- Assessment methods that have been or are used to evaluate the effectiveness of risk controls.
The how?
When developing the URRA, manufacturers should consider all intended uses of the product, the potential product users, and the likely environments of use. Each of these factors can impact the product design, user tasks, and the resulting risks and potential harms associated with the use of the product. The sequential steps of URRA development are summarized in Figure 1.
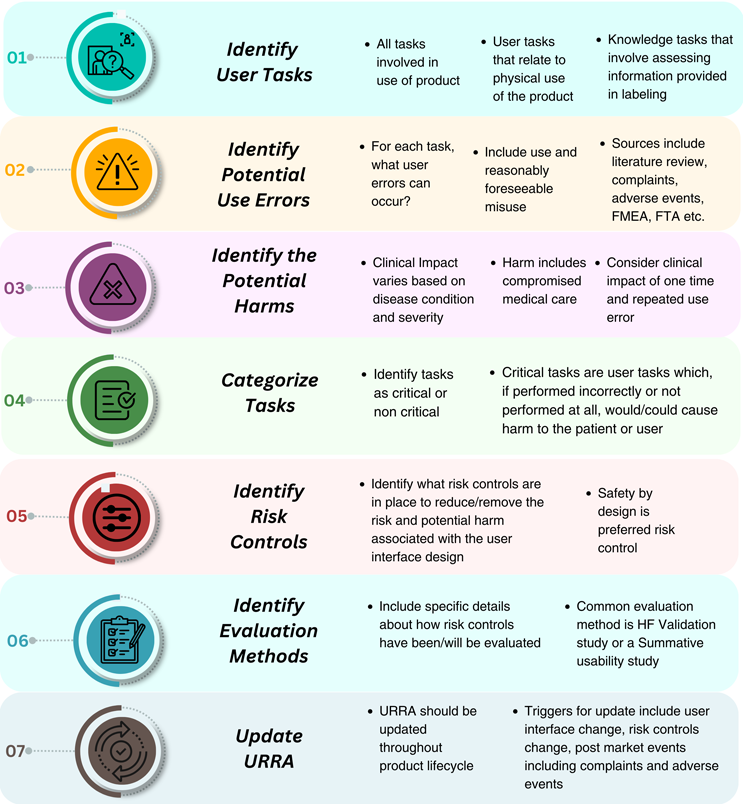
figure 1: URRA development process according to FDA guidelines.
Anatomy of a URRA
In Figure 2 we analyze the example given by the FDA in the guidance. The philosophy is simple and sequential, as detailed in Figure 1. The columns of the table deal in sequence with the following:
- Task number: A task identifier for traceability
- Description of the user task: Description of instructions for the user, e.g. wording to be included in the instructions for use (IFU)
- Potential hazards/clinical harm and severity: Describe the potential hazards and harms resulting from the application error and its associated severity from the perspective of clinical impact.
- Critical task: A user task is critical if it would or could cause harm to the patient or user if performed incorrectly or not at all. Critical tasks are most likely to be subjected to RF validation testing.
- Risk control measure: For each application failure, it is necessary to identify a risk control measure. This can be a feature that makes the design safe, a protective measure or safety information.
- Evaluation method: The assessment method should verify the effectiveness of the risk control measures. This is largely done through RF validation testing.
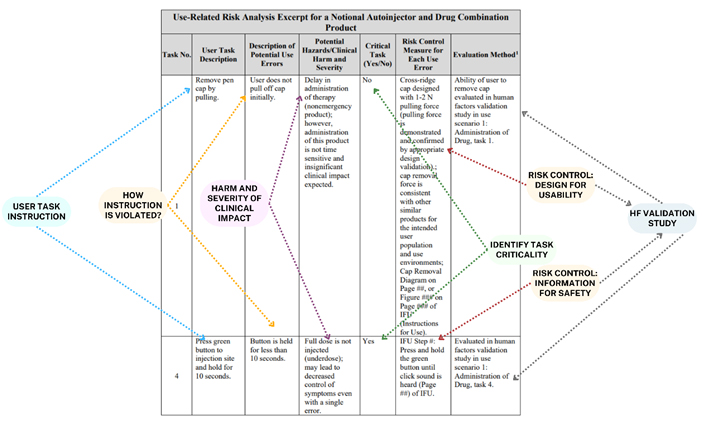
figure 2: Anatomy of URRA (FDA example).
comment
- While FDA usability guidelines, including this one, often align well with the AAMI HE75 standard, these guidelines still do not fully comply with the requirements of IEC 62366. This creates redundant documentation requirements for manufacturers seeking both FDA clearance and CE marking. For example, 62366 requires the identification of hazard-based use scenarios – something that is not in the FDA’s vocabulary.
- Column 4 in the URRA example in the guidance tries to cram too much into one column. It is better to split it into three columns that identify hazard, harm and severity separately.
- URRA is defined as a risk analysis tool. Risk includes probability of occurrence and severity. Where does URRA come in? Is it an FMEA? Is it a hazard analysis? Some guidance on how probability of occurrence and severity from safety risk management tools and methods are linked to URRA would have been helpful.
- The definition of harm in this guidance is broad and includes compromised medical care. This is where clinical impact and severity come into play to identify critical tasks. Although it is reasonable to assume that any significant clinical impact can be a critical task, manufacturers must agree internally on what is “significant” based on their severity scales according to the risk acceptance criteria.
- The last column in URRA is now labelled ‘Evaluation Method’. In some other previous guidelines it was called ‘Validation Method for the Effectiveness of Risk Mitigation Measures’. While the latter more clearly referred to an RF study, it may open the possibility of including other elements as the guidelines in Section III F are more flexible. Finally, a validation review of the final design is not required for all submissions in the RF category.
- Previously, FDA used the term “risk reduction” but now uses “risk control.” This is likely to align with ISO 14971:2019.
- While the title of the guideline mentions drugs and biologics first and combination products last, it is largely focused on combination products. Some more detail and nuance in the creation of the URRA for the former would have been welcome. We believe that the final guideline could incorporate this change, however small.
The public comment period ends on September 9, 2024.
About the authors:
 Jayet Moon holds a Masters degree in Biomedical Engineering from Drexel University in Philadelphia and is a Project Management Institute (PMI) Certified Risk Management Professional (PMI-RMP). Jayet is also a Chartered Quality Professional in the UK (CQP-MCQI). He is also an Enterprise Risk Management Certified Professional (ERMCP) and a Risk Management Society (RIMS) Certified Risk Management Professional (RIMS-CRMP). He is a Fellow of the International Institute of Risk & Safety Management. His new book, Basics of quality risk managementwas recently published by ASQ Quality Press. He holds ASQ CQE, CQSP and CQIA certifications.
Jayet Moon holds a Masters degree in Biomedical Engineering from Drexel University in Philadelphia and is a Project Management Institute (PMI) Certified Risk Management Professional (PMI-RMP). Jayet is also a Chartered Quality Professional in the UK (CQP-MCQI). He is also an Enterprise Risk Management Certified Professional (ERMCP) and a Risk Management Society (RIMS) Certified Risk Management Professional (RIMS-CRMP). He is a Fellow of the International Institute of Risk & Safety Management. His new book, Basics of quality risk managementwas recently published by ASQ Quality Press. He holds ASQ CQE, CQSP and CQIA certifications.
 Arun Mathew works in Quality Systems R&D at AbbVie. He holds an Executive MBA, an MSc in Real-Time Embedded Systems, and a Diploma in Electronics. He has over twenty years of experience in the medical device industry, drug development, manufacturing, and regulatory affairs. He is a long-time member of the American Society for Quality and is a certified CQA Certified Quality Auditor and CQE Certified Quality Engineer. Mathew has previously worked at Fortune 500 companies such as Zimmer Biomet, Medtronic, and Baxter.
Arun Mathew works in Quality Systems R&D at AbbVie. He holds an Executive MBA, an MSc in Real-Time Embedded Systems, and a Diploma in Electronics. He has over twenty years of experience in the medical device industry, drug development, manufacturing, and regulatory affairs. He is a long-time member of the American Society for Quality and is a certified CQA Certified Quality Auditor and CQE Certified Quality Engineer. Mathew has previously worked at Fortune 500 companies such as Zimmer Biomet, Medtronic, and Baxter.
